PACCMIT–CDS
Finding microRNA targets in the coding region is difficult due to the overwhelming signal encoding the amino acid sequence. PACCMIT–CDS [1] is an algorithm, which finds potential microRNA targets within coding sequences (CDS) by searching for conserved motifs that are complementary to the microRNA seed region and also overrepresented in comparison with a background model [2,3] preserving both codon usage and amino acid sequence. Thanks to the properly constructed background, the new algorithm achieves a lower rate of false positives and better ranking of predictions than do currently available algorithms, which were designed to find microRNA targets within 3′UTRs.
References:
- Marín RM, Šulc M, Vaníček J. 2013. RNA 19: 467 – 474.
- Fuglsang A. 2004. Biochem. Biophys. Res. Commun. 316: 755 – 762.
- Robins H, Krasnitz M, Barak H, Levine AJ. 2005. J. Bacteriology 187: 8370 – 8374.
PACCMIT
In summary, PACCMIT [1] is a flexible algorithm that filters potential miRNA binding sites in 3'UTR regions by their conservation, accessibility, or both, and then ranks the predictions according to the over-representation with respect to a random background based on a Markov model.
The ranking of the conserved target predictions according to the over-representation of the oligomer sequences in the 3'UTR complementary to the microRNA seed was proposed by Robins and Press [2] and used by Murphy et al. [3] to discover the roles of microRNAs in the reactivation and latency of herpesviruses. The PACMIT algorithm [4] applies this ranking only to partially accessible complementary sites, i.e., sites that contain a four–mer that appears in a single-stranded RNA region of at least 20% of the Boltzmann ensemble of secondary RNA structures. This accessibility filter was motivated by a method used by Robins, Li, and Padgett [5] for incorporating RNA structure to the target prediction. Marín and Vaníček [4] showed that filtering predictions by accessibility and ranking them by over-representation results in much higher precision than that obtained by other free–energy based methods.
Finally, the flexible algorithm that we call PACCMIT (Prediction of ACcessible and/or Conserved MIcroRNA Targets) [1], was obtained by combining PACMIT with a general conservation filter.
References:
- Marín RM, Vaníček J. 2012. PLoS ONE 7: e32208.
- Robins H, Press WH. 2005. Proc. Natl Acad. Sci. USA 102: 15557 – 15562.
- Murphy E, Vaníček J, Robins H, Shenk T, Levine AJ. 2008. Proc. Natl Acad. Sci. USA 105: 5453 – 5458.
- Marín RM, Vaníček J. 2011. Nucleic Acids Research 39: 19 – 29.
- Robins H, Li Y, Padgett RW. 2005. Proc. Natl Acad. Sci. USA 102: 4006 – 4009.
Unless stated otherwise, PACCMIT [1] is assumed within this web-site for predicting targets in the 3'UTR regions.
Contact
If you have any questions, please contact us either by e-mail or visit the web-page of our group, Laboratory of Theoretical Physical Chemistry, at the Ecole polytechnique fédérale de Lausanne (EPFL).
- E-mail jiri.vanicek@epfl.ch
- WWW http://lcpt.epfl.ch
Predictions
PACCMIT-CDS [1]
How are the resulting predictions ranked?
The PACCMIT–CDS algorithm is based on the assumption that functional binding sites that have avoided deleterious mutations should be overrepresented with respect to the composition of the surrounding sequence. Let $c$ denote the number of conserved seed matches (i.e., sites complementary to the seed sequence) in a given coding sequence and for a given microRNA. PACCMIT–CDS ranks its predictions according to an approximate probability $P_{\text{SH}}$ that a randomly generated sequence (based on a model preserving both the amino acid sequence and codon usage) would contain at least $c$ conserved seed matches. As usual, lower values of $P_{\text{SH}}$ suggest that the interaction is more likely to be functional.
How do you define conservation?
We employ the “Any–species” approach used by Marín and Vaníček [2]: a seed match is considered conserved if it is present in the aligned sequences of at least $S=12$ species (including the reference), regardless of their phylogenetic distance from the reference.
How did you assess precision and/or sensitivity?
Precision and sensitivity of PACCMIT–CDS were evaluated using PAR-CLIP and proteomics data sets. See Section Data sets of validated targets in [1].
References:
- Marín RM, Šulc M, Vaníček J. 2013. RNA 19: 467 – 474.
- Marín RM, Vaníček J. 2012. PLoS ONE 7: e32208.
PACCMIT [1]
How does PACCMIT score microRNA—3'UTR interactions?
The PACCMIT algorithm [1] is based on the assumption that functional binding sites that have avoided deleterious mutations should be overrepresented with respect to the composition of the surrounding sequence. Let $c$ denote the number of conserved and/or accessible seed matches (i.e., sites complementary to the seed sequence) in a given 3'UTR for a given microRNA. PACCMIT ranks its predictions according to an approximate probability $P_{\text{SH}}$ that a randomly generated sequence (based on a Markov model) would contain at least $c$ conserved and/or accessible seed matches. As usual, lower values of $P_{\text{SH}}$ suggest that the interaction is more likely to be functional.
What is the effect of the various filters (accessibility/conservation) within PACCMIT?
We showed [1] that while the conservation filter is more effective than the accessibility filter for predicting targets of highly conserved microRNAs, accessibility performs better than conservation in the case of weakly conserved microRNAs. Moreover, in the case of highly conserved microRNAs, the performance is improved even further, especially among the top predictions, by using the combined filter.
How do you evaluate accessibility within PACCMIT/PACMIT?
The algorithm for calculating accessibility is common to both PACMIT [2] and PACCMIT [1]. A 7–mer in the 3'UTR sequence is catalogued as accessible if it contains at least one 4–mer located in the single stranded region of at least 20% of RNA secondary structures. The probability of a 4–mer to appear in a single–stranded region is evaluated with the program RNAplfold [3] using a window $W=80$ and a maximum pairing distance $L=40$ [4].
How do you treat conservation in PACCMIT?
The conservation filter in PACCMIT was optimized using a training dataset constructed from the proteomics data by Baek et al. [5] and Selbach et al. [6]. We computed [1] precision and the number of true targets per microRNA as functions of the number of predictions per microRNA for varying stringency of the conservation filter and found that the conservation filter with four selected species (human, chimp, rhesus, mouse) outperformed the filter with any twelve species and thus was used by default in all PACCMIT analyses.
References:
- Marín RM, Vaníček J. 2012. PLoS ONE 7: e32208.
- Marín RM, Vaníček J. 2011. Nucleic Acids Research 39: 19 – 29.
- Bernhart SH, Hofacker IL, Stadler PF. 2006. Bioinformatics 22: 614 – 615.
- Tafer H, Ameres SL, Obernosterer G, Gebeshuber CA, Schroeder R, et al. 2008. Nat. Biotechnol. 26: 578 – 583.
- Baek D, Villén J, Shin C, Camargo F, Gygi S, et al. 2008. Nature 455: 64 – 71.
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, et al. 2008. Nature 455: 58 – 63.
Tutorial
The purpose of this short tutorial is to demonstrate a typical usage of this web server. The recipe below guides the user through the fours steps of the wizard accessible via the link Predictions in the main menu.
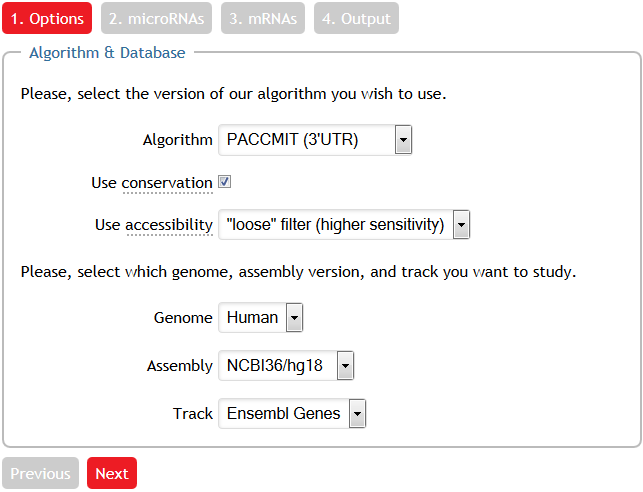
Choosing the algorithm & database
In our example, we select the PACCMIT algorithm and invoke the conservation and accessibility filters. Also, note that we explictly request Ensembl genes.
Selecting microRNAs of interest
Let us assume that we are interested in the targets of the microRNAs listed in the figure below. In general, the accession numbers (not the IDs) can be pasted in the window by hand, but the specific microRNA accession numbers shown in the figure can be loaded automatically by clicking the Load sample data button.

Selecting mRNAs of interest
In this step, we specify the mRNAs. Since we selected Ensembl genes, we have to paste the identificators in the corresponding format. As in step 2, the specific set of mRNA IDs shown in the figure can be loaded automatically by clicking the Load sample data button.

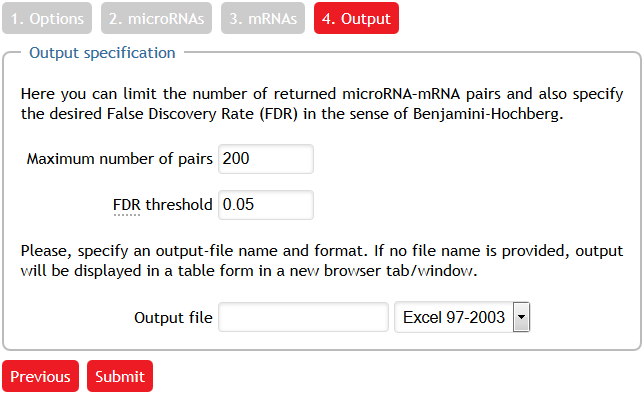
Imposing output parameters
Finally, we impose that we are interested in at most 200 microRNA—mRNA pairs. However, this constraint does not apply in our example since the maximum possible number of returned microRNA—mRNA pairs is $3\times4=12$. Moreover, we set the (Benjamini-Hochberg) FDR threshold to 5%. Since the input field Output file is left empty, the output is redirected into a new browser window.
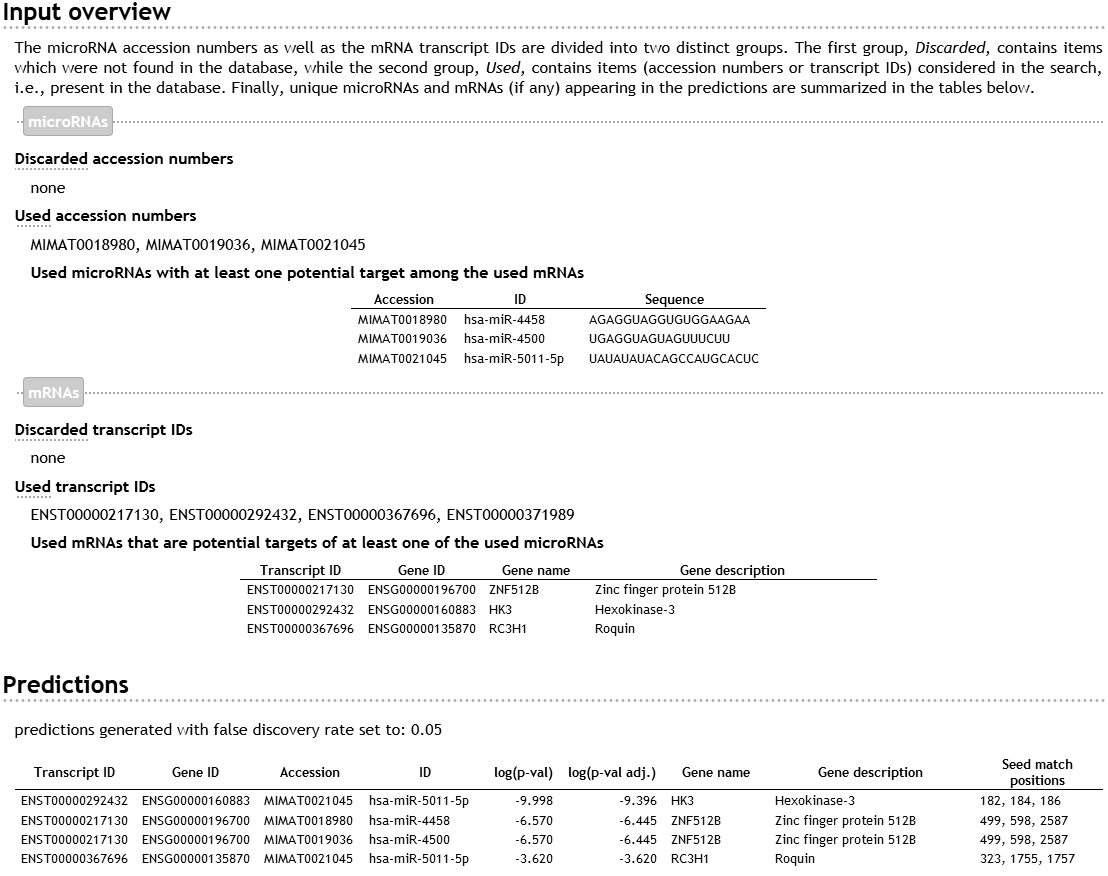
Structure of the output
The top part of the output, labeled as “Input overview,” shows which user-supplied miRNA accession numbers and mRNA IDs were discarded and which were used in the search. The top section also explicitly lists those used miRNAs from the input that have at least one potential target among the used mRNAs and those used mRNAs from the input that are potential targets of at least one used miRNA. By a potential target is meant a mRNA that contains a seed match (i.e., a 7-mer complementary to the seed of a given miRNA) satisfying the required accessibility and/or conservation filters. The web server automatically provides IDs and nucleotide sequences for the miRNAs as well as gene names/descriptions for mRNAs.